Abstract
Primary diffuse meningeal melanomatosis (PDLM) represents a rare and aggressive form of primary central nervous system (CNS) melanoma, with only 32 cases reported in the literature [1]. Given its rarity, the clinical and biological behavior of PDLM has not been extensively studied, and standardized management protocols are lacking. The disease carries a poor prognosis, with the literature reporting a median survival rate of only 7 months. We present the case of a 7-year-old boy who exhibited symptoms of raised intracranial pressure and fever, which mimicked an infective etiology but was ultimately
Background
Primary diffuse meningeal melanomatosis (PDLM) represents a rare and aggressive form of primary central nervous system (CNS) melanoma, with only 32 cases reported in the literature [1]. Given its rarity, the clinical and biological behavior of PDLM has not been extensively studied, and standardized management protocols are lacking. The disease carries a poor prognosis, with the literature reporting a median survival rate of only 7 months. We present the case of a 7-year-old boy who exhibited symptoms of raised intracranial pressure and fever, which mimicked an infective etiology but was ultimately
Case summary
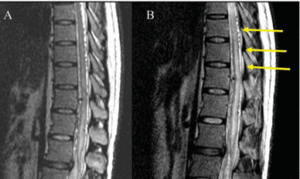
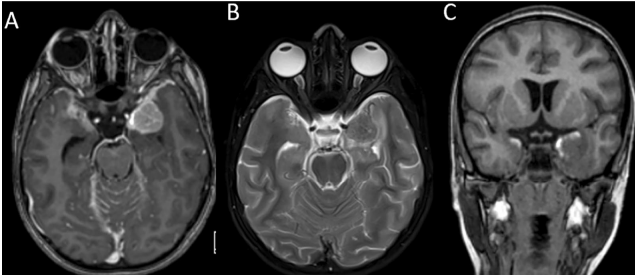
A 7-year-old child from North India with no significant past medical illness presented to the pediatric casualty with a 15-day history of fever, headache, projectile vomiting, and loss of appetite. On examination, bilateral papilledema was noted and there were no neurological deficits. He was admitted to the pediatric ward with suspected meningitis, probably of tubercular origin. Given the child’s symptoms of raised intracranial pressure, a lumbar puncture was deferred, and a CT brain scan was performed. The scan revealed a well-defined round hyperdensity in the left middle cranial fossa along the lateral wall of the cavernous sinus and the greater wing of the sphenoid which appeared extra-axial. An MRI scan (Fig. 1) displayed diffuse leptomeningeal enhancement of the brain and spine, accompanied by a small enhancing subpial nodule in the left medial temporal lobe, showing faint T1 hyperintense areas within it. Thick
leptomeningeal enhancement was noted along the bilateral sylvian fissures, temporal convexities, occipital sulci, cerebellar folia, ventral brainstem, and upper cervical cord. The subpial nodule measured 2.2×2.1×2.0 cm (TR x AP x CC) and was T2 hypointense. After clinicoradiological correlation, potential differential diagnoses were considered including infectious, neoplastic or inflammatory conditions. Consequently, an open micro-neurosurgical biopsy was scheduled. A left temporal craniotomy was performed, and the left temporal subpial nodule was biopsied. Intraoperatively, diffuse black staining of the arachnoid was noted along the sylvian fissure. The subpial nodule in the left medial temporal region was black in color, firm in consistency, and had necrotic areas within. The M2 branches were located within the lesion. Upon frozen-section analysis, histopathologists identified oval to plasmacytoid atypical cells with prominent nucleoli and cytoplasmic brownish pigment, suggesting a melanocytic lesion. Subsequent histopathological sections (Fig. 2) revealed a tumor composed of sheets of polygonal and spindle-shaped cells with hyperchromatic, eccentrically placed nuclei, some of which had distinct nucleoli and moderate amounts of eosinophilic cytoplasm. Mitotic activity was evident, and many tumor cells contained intracytoplasmic brown granular pigment, which tested positive for immune staining with anti-melanosomal antibody MART-1 (Melan-A) and bleached when exposed to melanin bleach. Small foci of necrosis were also present. On immunohistochemistry, the tumor was positive for HMB-45, faintly positive for CD-117, and retained expression of INI-1. MIB-1 labeling index was 55%. BRAF mutation was negative. The biopsy was reported as a malignant melanocytic tumor. A whole-body PET-CT ruled out a primary lesion elsewhere in the body. The patient had no cutaneous or eye lesions. Radiologic and histologic findings and the absence of cutaneous lesions led to a diagnosis of PDLM (Table 1). Two weeks after discharge, he was readmitted to the hospital due to recurring headaches, accompanied by multiple episodes of vomiting and one instance of abnormal posturing. A CT scan detected hydrocephalus, necessitating the placement of a right parietal mediumpressure ventriculoperitoneal shunt. Following the procedure, his symptoms resolved. Cerebrospinal fluid cytospin analysis revealed scattered atypical cells with enlarged nuclei, coarse, dark, granular chromatin, and prominent nucleoli. He subsequently received adjuvant craniospinal irradiation with a left temporal boost with palliative intent. A dose of 3000 cGy in 10 fractions was administered to the brain and spinal cord, while a boost of 1500 cGy in 5 fractions was given to the residual left temporal lesion. He received adjuvant temozolomide. He expired 2 months after his treatment was completed. Review of literature According to the 2021 WHO classification, primary CNS melanocytic neoplasms are classified as diffuse meningeal melanocytic neoplasms and circumscribed meningeal melanocytic neoplasms [2]. Diffuse meningeal melanocytic neoplasms are further classified as meningeal melanocytosis and meningeal melanomatosis. In meningeal melanocytosis, tumor cells look bland and do not infiltrate the brain, while in meningeal melanomatosis (PDLM), the tumor cells have marked atypia, mitotic activity, and infiltration into the brain [2].